2022年岁末,伴随一声啼哭响彻整个产房,一个健康新生命诞生了。
这一幕的背后是我院医学遗传与产前诊断中心联合多学科,为一个被罕见病困扰四代人的家庭找到病因,家庭成员安心迎接健康新生命的故事。
查阅检测报告,发现多个致病可能性
据了解,20多年前,黄女士(化名)的父亲及两位叔叔都在风华正茂的年纪相继出现行走不稳、手脚无力等症状。随后,黄女士的父亲和其中一位叔叔不幸因病去世,而另一位叔叔目前已生活不能自理。黄女士的奶奶及其他长辈也曾有类似症状,虽然黄女士尚未出现身体异常,但她十分担忧自己今后是否会发病以及疾病是否会持续影响下一代,为此,黄女士四处求医,进行了相关基因检测,但都未能找到病因。
为了能继续解开困惑,消除疑虑,9个月前,黄女士来到我院医学遗传与产前诊断中心就诊。接诊医师仔细查阅了一份2年前黄女士和她的叔叔及姑姑在外院做的全外显子组测序+多重连接探针PCR(MLPA)进行的脊髓性肌萎缩症基因诊断+运动神经元存活基因1(SMN1)检测报告。
这长达18页的基因检测报告提示1个与临床症状相关的高度可疑变异:杜氏肌营养不良(DMD)基因突变。接诊医师还发现了4个较为可疑的变异:涉及运动神经元存活基因2(SMN2)的7、8号外显子杂合缺失、运动神经元存活基因1(SMN1)的错义突变、肌联蛋白基因(TTN)以及亲联蛋白1基因(JPH1)的错义突变。同时还有4个与肌病相关的变异。
多个致病可能性,谁是最后的“真凶”呢?
多学科联合,循迹查出“真凶”
由于疾病涉及家族四代人中的多个家庭成员,医学遗传与产前诊断中心医师通过患者出现的行走不稳、手脚无力等症状,认为可能会涉及肌肉、神经等萎缩导致的疾病。经过详细问诊,医师为该家族绘制出家系图。
家系图
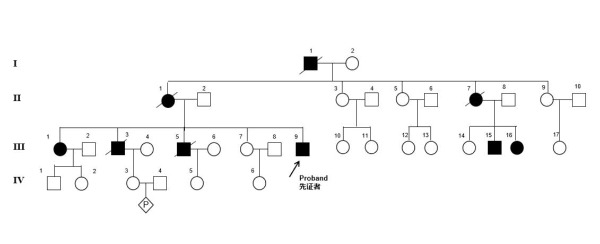
符号说明:“□”代表正常男性,“■”代表患病男性,“◯”代表正常女性,“●”代表患病女性,“◇”代表胚胎或胎儿,“┰”代表婚姻及生育后代,“↗” 箭头指向为先证者(即家系中第一个进行检查和诊断的个体),“/”代表个体死亡;“I、II”等罗马数字代表家族中第几代,阿拉伯数字代表每代中个体的顺序。
家系图显示,四代人中有三代人受累及,成年发病、进行性加重,男女均出现发病患者,家族中正常的子代所生育的后代均正常,而患者与正常人婚配,后代既有患者也有正常人。根据这个遗传规律,医师初步判断此病为常染色体显性遗传疾病可能性大,诊断的目标进一步缩小。但要查出“真凶”,单纯凭借家系图提供的信息还远远不够。
由于年代久远,家族中部分患者的体格检查、化验及影像学、神经系统专科资料已经无法查找。在此情况下,大部分咨询者往往会选择放弃,但是黄女士在家人的支持下坚持继续寻求真相。
为此,医学遗传与产前诊断中心联合神经内科专家组成团队,对黄女士罹患疾病的叔叔(箭头所示先证者)进行了体格检查、实验室的肝酶及肌酸激酶检测以及肌电图和大脑核磁共振等检查。在排除了脊髓性肌萎缩症(SMA)、肌营养不良相关疾病及脑白质病变的可能性后,对疾病诊断的方向进一步指向遗传性脊髓小脑共济失调(spinocerebellar ataxia,SCA)。
由于遗传性脊髓小脑共济失调主要是因为其致病基因编码区内三核苷酸CAG重复序列异常扩增(动态突变)。而目前基于二代测序的全外显子测序技术不能诊断动态突变,这就是黄女士和她的叔叔及姑姑在外院做的相关检测没有检测出病因的原因。
正是有了前面的遗传咨询和家系图分析以及相应基因检测报告作为排除的依据,医学遗传与产前诊断中心医师为黄女士和她的叔叔选择了能够进行CAG动态突变的基因检测方案,经检测确诊为脊髓小脑共济失调3型的致病基因(ATXN3)三核苷酸CAG动态突变数目异常。可喜的是黄女士不携带该致病基因的动态突变。
“从早孕的忐忑不安,到孕中期明确自己未携带致病基因的动态突变,到最后放下心里的担忧,感谢医院的医师们给了我这份安心。”问诊期间,黄女士怀孕,并在2022年岁末产下了健康的宝宝。
罕见病的诊断需要有精准的检测手段,同时也需要患者意识到筛查的重要性。开展早期筛查是预防罕见病的有利手段,我院医学遗传与产前诊断中心专家建议,大家要重视基因检测,预防罕见病,及早发现,及时干预。
知识拓展
遗传性脊髓小脑共济失调是一类罕见的小脑性共济失调疾病,遗传方式以常染色体显性遗传为主,其他遗传方式(常染色体隐性、X-连锁遗传)很少见。因患者中枢神经系统的小脑、脑干和脊髓均受累,故称为脊髓小脑性共济失调。
本病发病率约为3/100000,是一种慢性进行性家族的退行性疾病。该疾病的症状主要为成年发病,随着患者的小脑逐渐退化,脑干和中枢神经系统的其他部分也会发生退行性改变,从而表现为共济失调、眼球运动障碍、构音障碍、肌张力障碍及眼球震颤等。目前有超过30种亚型被报道,每个亚型都有共济失调的症状,但次级症状差别很大。(覃婷 文/图)
参考文献
1.邬玲仟,梁德生. 《人类单基因遗传疾病》〔M〕.西安交通大学出版社.
2.Klockgether T. The clinical diagnosis of autosomal dominant spinocerebellar ataxias〔J〕.Cerebellum,2008,7(2):101-105.
3.Perlman S L. Ataxias〔J〕. Clin Geriatr Med,2006,22(4):859-877.
4.Smith D C, Bryer A, Watson L M,et al. Inherited polyglutamine spinocerebellar ataxias in South Africa〔J〕. S Afr Med J,2012,102(8):683-686.5.Paulson H L. The spinocerebellar ataxias〔J〕. J Neuroophthalmol, 2009,29(3):227-237.











 服务号
服务号
 订阅号
订阅号
 官方微博
官方微博
